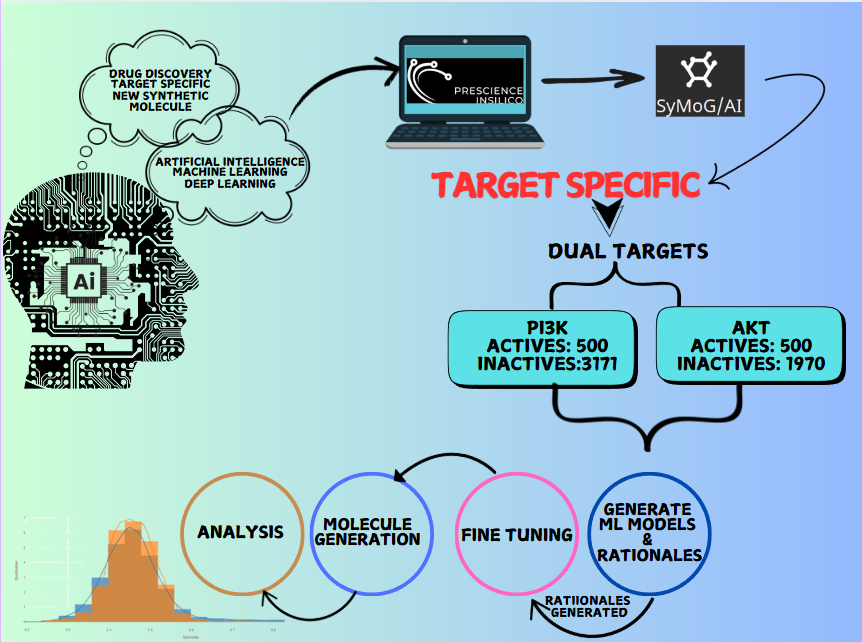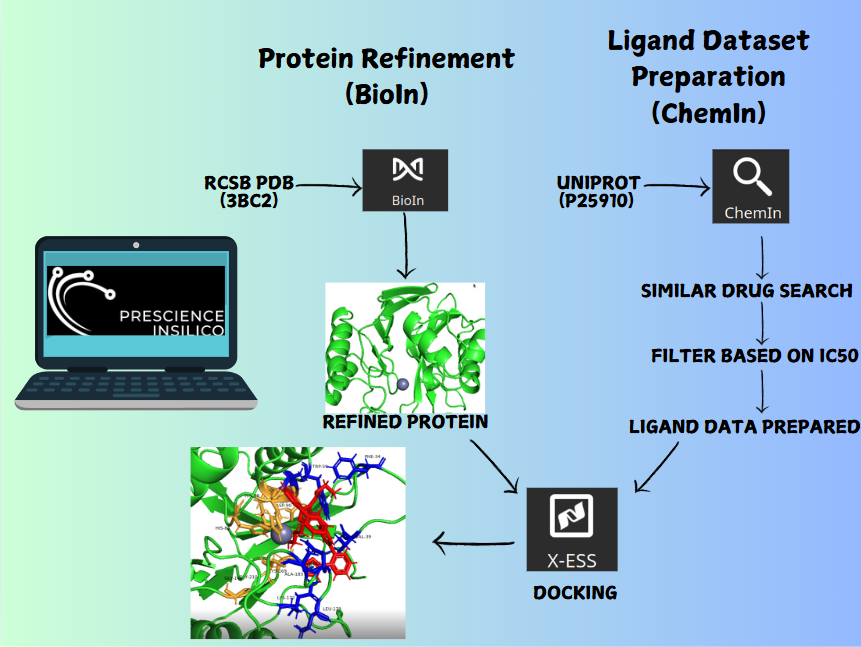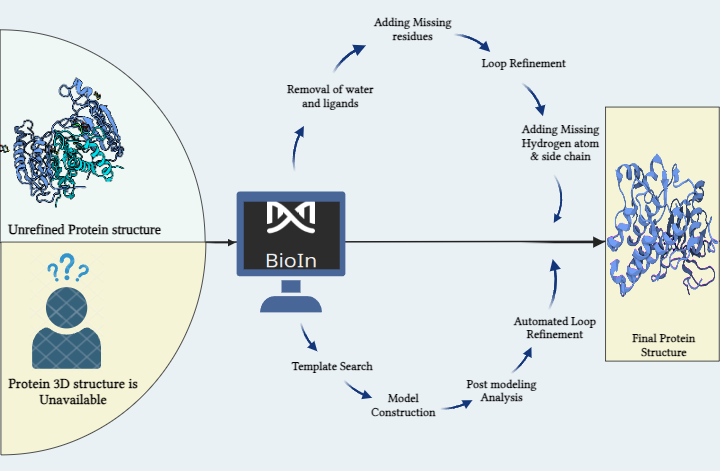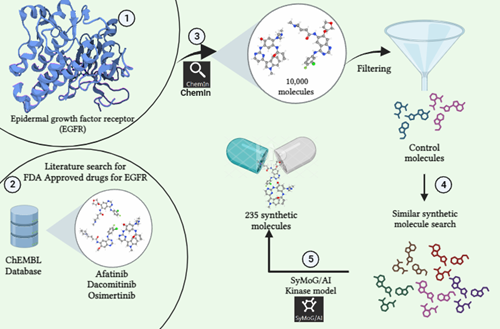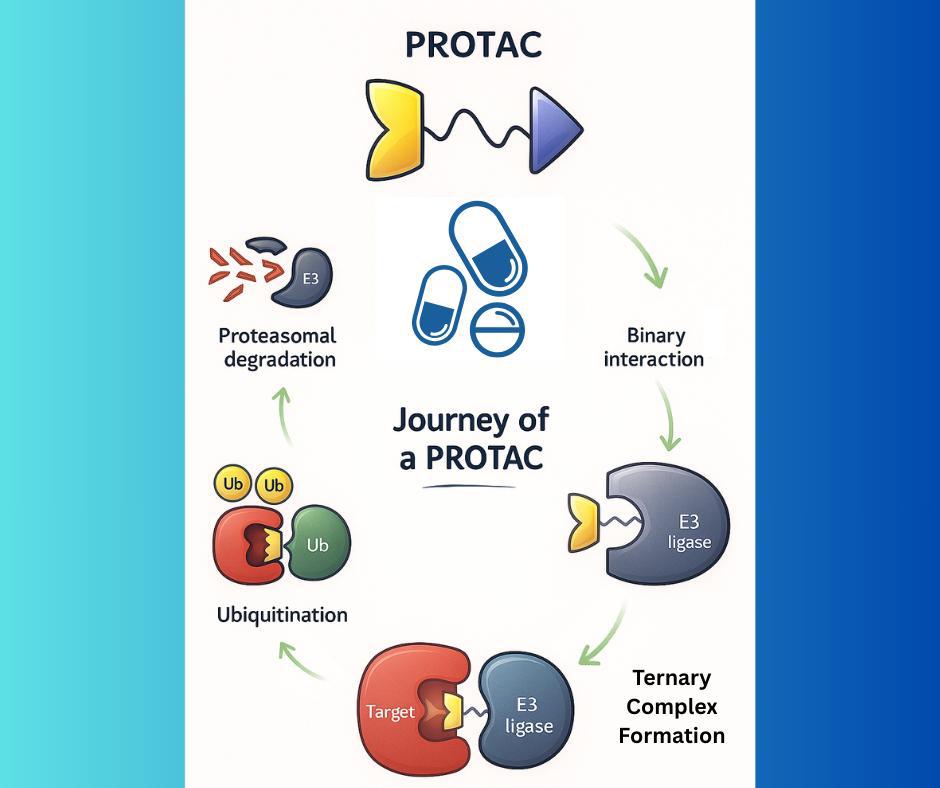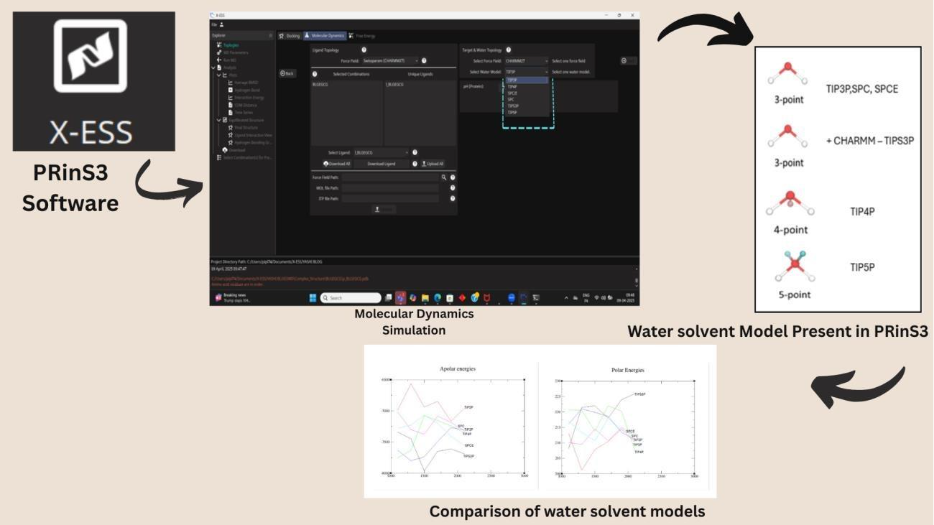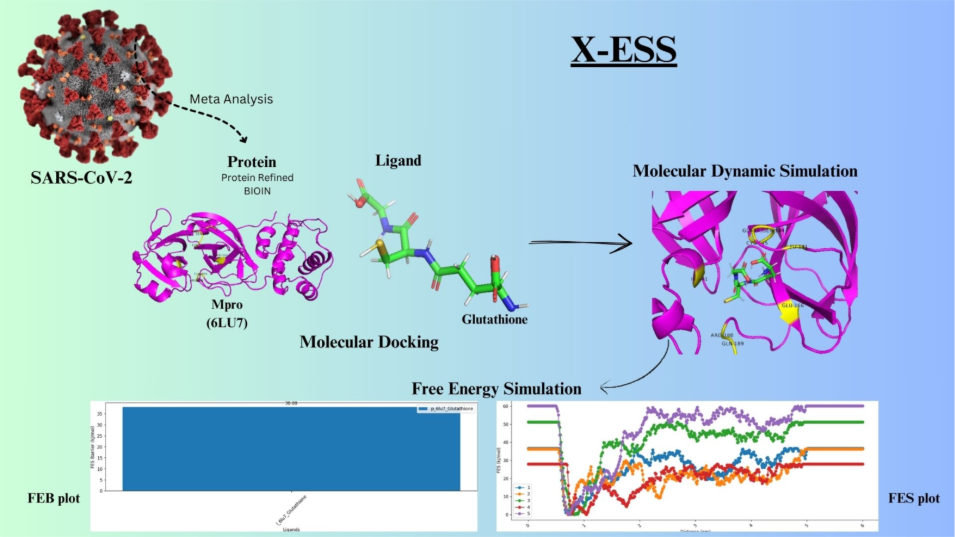
Metadynamics-Based Free Energy Simulations of Glutathione Binding to SARS-CoV-2 3CLpro
The COVID-19 pandemic, caused by the SARS-CoV-2 virus, has underscored the urgent need for effective antiviral therapeutics. One of the most promising drug targets is the 3C-like protease (3CLpro), also known as the main protease (Mpro), which plays a critical role in viral replication and transcription. Computational drug discovery approaches, such as molecular docking, molecular dynamics (MD) simulations, and free energy calculations, have become indispensable tools