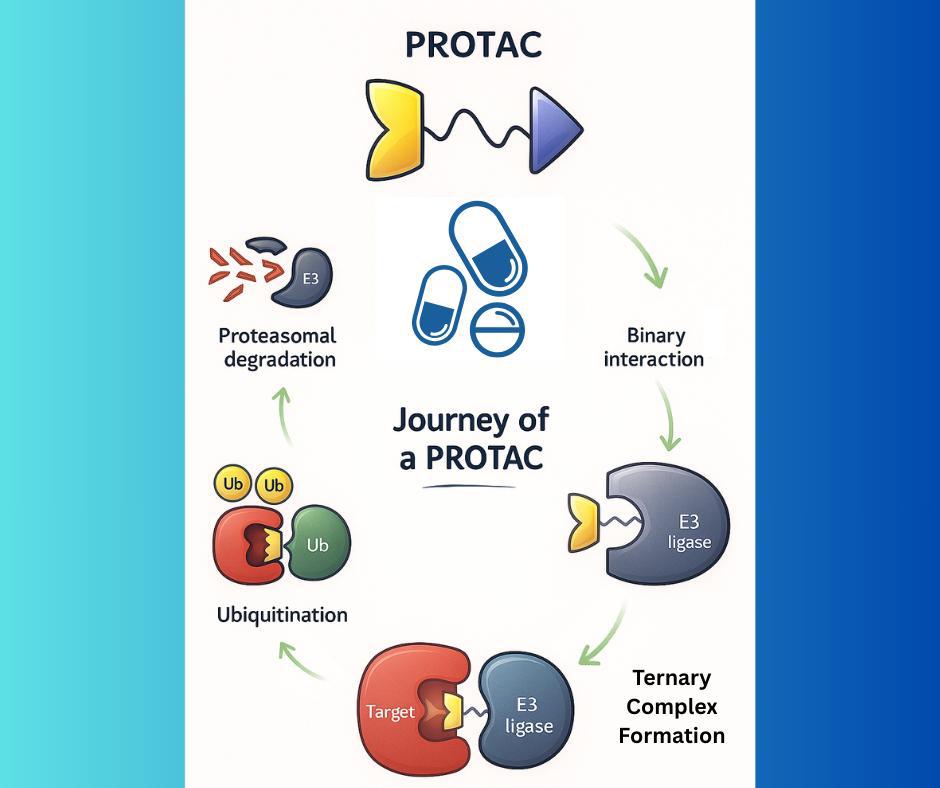
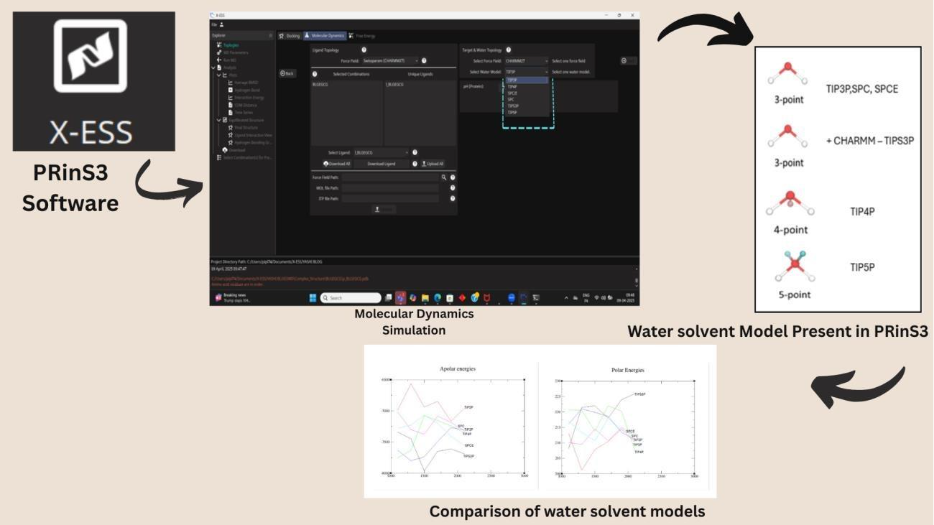
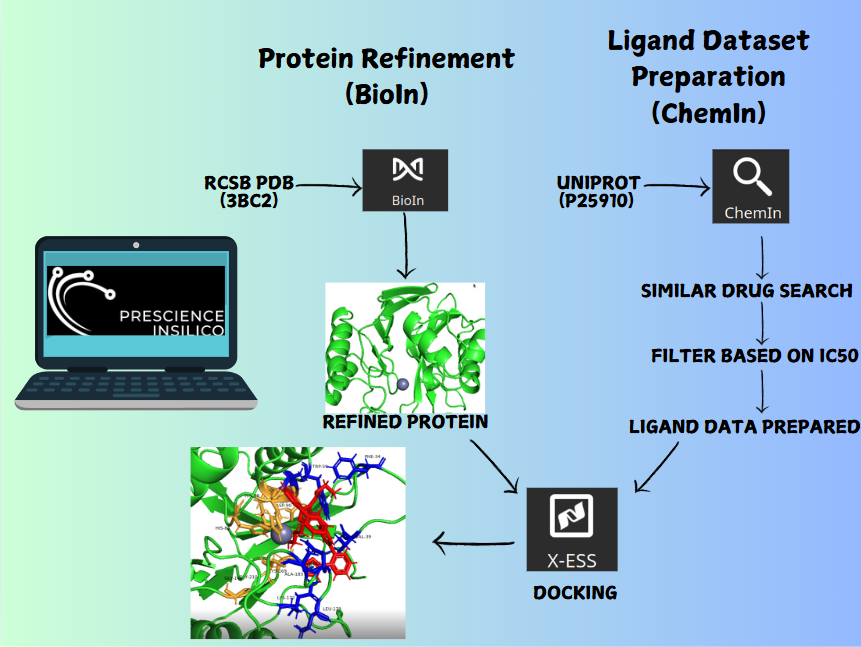
Metadynamics-Based Free Energy Simulations of Glutathione Binding to SARS-CoV-2 3CLpro
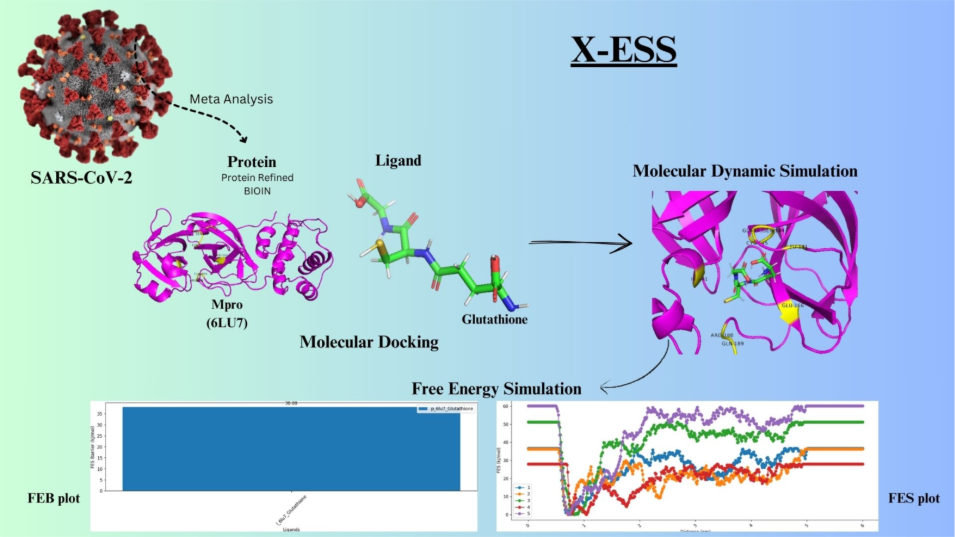
The COVID-19 pandemic, caused by the SARS-CoV-2 virus, has underscored the urgent need for effective antiviral therapeutics. One of the most promising drug targets is the 3C-like protease (3CLpro), also known as the main protease (Mpro), which plays a critical role in viral replication and transcription. Computational drug discovery approaches, such as molecular docking, molecular dynamics (MD) simulations, and free energy calculations, have become indispensable tools for identifying and optimizing potential inhibitors. This case study demonstrates how the XESS application within the PRinS3 software was utilized to perform Free Energy Surface (FES) simulations and metadynamics-based ligand unbinding free energy calculations to identify and optimize potential 3CLpro inhibitors. The study aimed to evaluate glutathione as a potential peptide inhibitor by integrating docking, MD simulations, and free energy analysis.
Simulation-Based Evaluation of 3CLpro–Glutathione Complex Stability
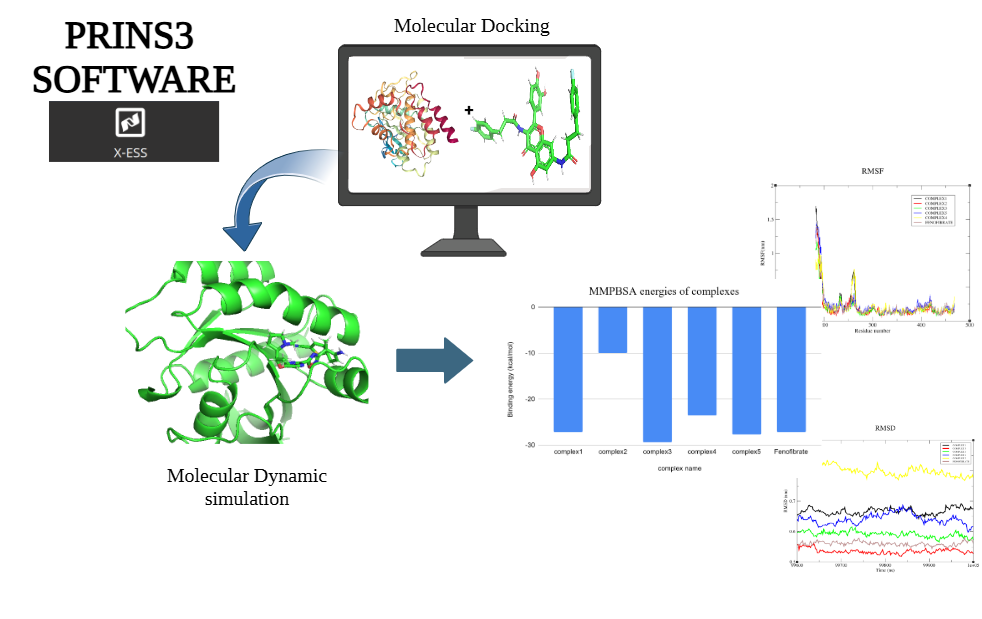
The study was initiated with molecular docking to predict the binding mode and affinity of glutathione within the 3CLpro active site. Docking results provided initial insights into the protein-ligand interactions, which were further refined using molecular dynamics (MD) simulations. MD simulations allowed for the exploration of the dynamic behavior of the 3CLpro-glutathione complex over time, revealing conformational changes, stability, and key interactions at an atomic level. Following MD simulations, the conformations of the protein and ligand were used as input for free energy simulations. To down-select the most promising ligand poses, a scoring system was developed based on MD results, incorporating parameters such as Root-Mean-Square Deviation (RMSD), Internal energy, and hydrogen bonding. These parameters were weighted according to user-defined priorities, providing a comprehensive evaluation of each ligand pose.
Free energy simulations were conducted using the Metadynamics enhanced sampling technique, implemented through the XESS application in PRinS3. Metadynamics is particularly effective for calculating dissociation free energies, as it allows for the exploration of rare events and energy barriers in protein-ligand systems. The simulation setup required the specification of PLUMED input parameters, including Gaussian height, width, reaction coordinate, bias deposition pace, bias factor, and output file printing stride. The reaction coordinate chosen for biasing the system was the distance between the centers of mass of the heavy atoms of glutathione and the 3CLpro binding pocket. Five independent free energy surface (FES) simulations were performed to ensure robust results, with users having the flexibility to adjust the number of simulations and parameters as needed.
Comprehensive Analysis of Protein–Ligand Binding
Upon completion of the simulations, HILLS and COLVAR files were generated, containing data on the free energy landscape of the system. These files were analyzed to plot free energy surfaces (FES) and determine the free energy barrier for glutathione dissociation from 3CLpro.
Overall, docking and MD simulations consistently indicated a favorable binding of glutathione to the 3CLpro active site, with MM-PBSA calculations (-27.2±2.1 kcal/mol) revealing a significantly stronger binding affinity compared to the initial docking score (-8.84 kcal/mol). Key hydrogen bonds with residues such as His163, Gly143, His164, Ser144, Cys145, Glu166, and Leu141 contributed to the stability of the complex. Furthermore, free energy surface analysis identified a dissociation barrier of 38.08 kcal/mol, suggesting that glutathione forms a stable and energetically favorable interaction with 3CLpro. This analysis offered valuable insights into the binding stability and dissociation kinetics of glutathione, underscoring its potential as a therapeutic candidate.
In conclusion, the integration of computational and experimental approaches provided a robust framework for evaluating glutathione as a potential COVID-19 treatment. The XESS application in PRinS3 software proved to be a powerful tool for conducting free energy simulations, enabling the estimation of binding affinities and the identification of key interactions for lead optimization. This case study demonstrates the potential of computational drug discovery in accelerating the development of antiviral therapies, offering a promising pathway for combating emerging infectious diseases.
Complete details about these applications can be found on our official website at https://prescience.in/. To obtain access to the platform and obtain all the information required for the best possible experience, we respectfully ask that you contact us by email at support@prescience.in and obtain information at https://prescience.in/prins3/.